-ADA总体发生率:治疗增强和治疗引起ADA阳性受试者的总和,除以可评估受试者的总数,而得出的百分比。不包括给药后没有任何样本供评估,基线阳性的受试者。
-治疗引起的ADA发生率:治疗引起ADA阳性受试者总数,除以可评估的,基线ADA阴性的受试者总数,而得出的百分比。此外,需要报告此组受试者滴度的峰值和范围(中位数、IQR)。
•中和性ADA:如果适用的话,按上文所述分析报告预先存在的NAb、增强和发生率。如果ADA在所有受试者中都是中和性ADA,则无需作单独的分析。
•ADA动力学:ADA出现的时机及其持续时间对于临床医生监测治疗的进展非常有用。抗体的持续性在几个病例中显示与临床效应相关联。
对药物开发人员而言,关于ADA动力学知识有助于优化同一生物药后续研究中的采样计划,以及作为药物上市后药物警戒计划的一部分,助力ADA监测计划的优化、风险的管理和缓解。
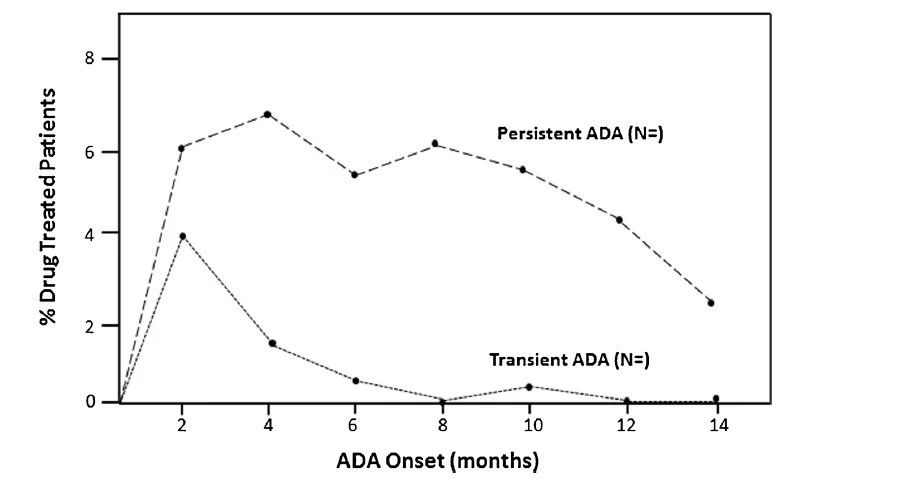
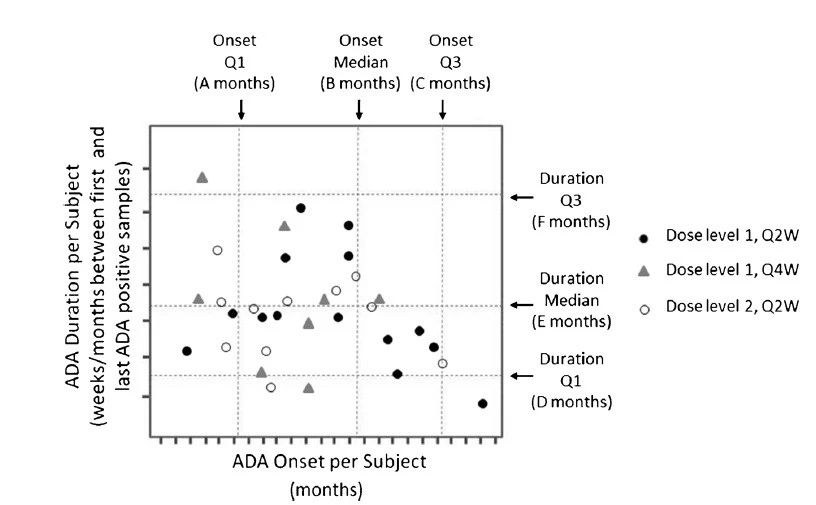
ADA动力学的图像表示是最有用的。例如,图1中说明了ADA开始和ADA持续时间的双变量图;图2中所示为瞬时态与持续性ADA频率图。当ADA阳性受试者数量较多(例如≥20),并且研究持续时间足够长,足以识别其发育后持久性抗体(例如≥1年)时,这些类型的图表蕴含的信息最丰富。
图1.治疗引起的ADA动力学: 发生和持续时间。ADA阳性结果持续时间与ADA发生时间的示意图。垂直和水平网格线绘制在分布的四分位数处:这有助于确定ADA的持续性或瞬时性是否与在患者中观察到ADA的时间相关。为了保证评估的准确性,只有那些ADA始发时间是上次访问前至少16周,或者在上次访问时或之前是ADA阴性的患者,才应列入此图。在解释此示意图时,应牢记在较晚的ADA始发时间的最大持续时间将按比例减少。图中的符号可以指示所选择的变量(本例中是剂量),临床效应,如:对疗效的影响(例如:是,否,和研究中断),不良反应(例如:是,否,和研究中断)等。
当样本量足够大时,还建议对结果进行更多的统计描述。这种客观的方法可以防止由于主观偏见而曲解结果。但值得注意的是,对于样本量大小的评估应根据具体情况判断,也要取决于临床研究的设计。建议采用以下计算方法:
(a)ADA的始发(Onset):指在研究中初次给药到发现第一例治疗引起ADA的时间段。使用实际经过的时间是计算该时间段的理想选择,不过使用最初设定的研究时间段也是可行的。计算“ADA 出现的时间中位值(median time to ADAdevelopment)”和四分位数 Q1 和 Q3,可以分别用来解释50%、25%和75%的ADA阳性受试者的ADA始发时间。分析ADA始发相关的其他参数可以是:“到ADA始发的给药次数"或 "到ADA始发的药物暴露天数”。
(b)ADA的持续时间:指药物引起的ADA的寿命。计算和报告诱导发生的ADA响应的中位持续时间和IQR,对于评估其与临床效果的相关性,是最客观的方法。但粗略地将 ADA 分类为瞬时性与持续性的方法占主导地位。虽然没有必要使用此类术语进行分类,但在应用这些术语时,使用统一定义就变得非常重要了。因为天然(内源性)的人类IgG1,IgG2和IgG4的半衰期大约在21-25天左右,五个半衰期大约等于16周。如果ADA只被药物诱导产生,并且从未被重新刺激或增强(一种"瞬时态"抗体),它将受人体的天然清除机制的约束。因此,ADA预计将在五个半衰期之后被完全清除(实际上只剩下微不足道的3%)。因此,可以用此现象区分瞬时性(血清返还sero-reverting)与持续性ADA,并建议用以下方法来评估ADA的持续时间:
• 瞬时性 ADA 响应:
–药物治疗引起的ADA在治疗或后续观察期间只在一个采样时间点检测到(不包括最后一个采样时间点,除非在之后被证明无法检测到),否则应视为持续性的,或:
–药物治疗引起的ADA在治疗期间(包括随访期,如果有的话)有两个或两个以上采样时间点被检测到,其中第一个和最后一个ADA阳性样本(无论中间有任何阴性样本)是小于16周的时间段间隔,并且受试者在最后一个采样时间点是ADA阴性。
• 持续性 ADA 响应:
–药物治疗引起的ADA在治疗期间(包括随访期)有两个或两个以上采样时间点被检测到,其中第一个和最后一个ADA阳性样本(无论中间有任何阴性样本)间隔有16周或更长,或:
–药物治疗引起的ADA发生率仅在治疗研究期的最后一个采样时间点,或者与上一个ADA阴性的间隔不到16周的采样时间点。虽然很少见,但如果IgG3或IgA的ADA在研究人群中占主导地位,则应用5周(而不是16周)的时间段来修改瞬时性和持续性ADA的定义。这是因为IgG3和IgA的半衰期比其他IgG短(IgG3为7天,IgM和IgA为5天)。
请注意,治疗增强的ADA被排除在ADA动力学分析之外,因为这种类型的免疫反应在机制上有所不同。在预先存在的ADA非常普遍的情况下,单独描述增强ADA的动力学可能很有用。这时,无需将ADA响应分为瞬时性(transient)和持续性响应。计算ADA的中位持续时间和四分位数(Q1和Q3)后就可以分别描述50%、25%和75%的ADA阳性受试者的ADA持续时间。四分位数方法可以更好地阐明ADA持续时间与临床效应之间的关系(如果有的话)。最后一点,将瞬时性和持续性抗体分别定义为在研究结束前消失和在最后研究时间点仍然存在的抗体,是不太合适的,这是因为瞬时性和持续性ADA的定义将取决于临床研究的长度,而并非ADA实际持续的时间。如果使用这样的定义,较长的临床研究会将ADA的性质偏颇地判断为"瞬时性抗体"。
• NAb 发生率和动力学:
当研究结果表明:受试者可以根据他们是否拥有NAb与non-NAb 而分组时,可以运用上文所述方式,分别详细考察每个组NAb的发生率和动力学。
• 交叉反应性:
当生物药物分子与内源性蛋白(全部或部分)相同或几乎相同时,评估ADA与内源性蛋白的交叉反应性非常重要,因为人们越来越担心这种ADA可能导致以内源性蛋白质耗尽为特征的自身免疫性综合征。将具有交叉反应性的ADA和对药物分子的ADA滴度和动力学进行比较会有助于评估相关疾病的恶化。
图2.治疗引起的ADA发生动力学: 一个研究实例中的瞬时和持续ADA免疫反应的发展。每一点表示在所示发病时间出现ADA的受试者的百分比,其持续时间可能是短暂的或持久的。在此示例中,10%的受试者有 2个月的ADA始发时间,其中4%具有瞬时性ADA响应,6%具有持续性 ADA响应。类似的,在6个月的ADA始发时间,0.5%有瞬态ADA响应,5.5%有持续的ADA响应。该图的横轴也可以使用剂量。
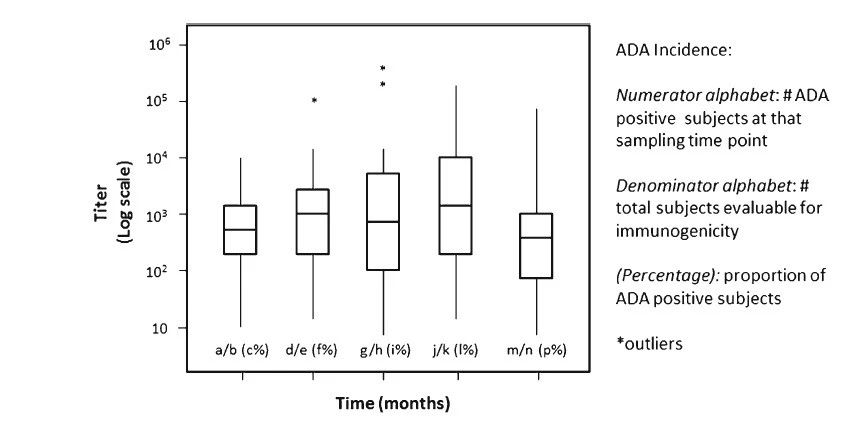
可以使用替代方法描述这些ADA的属性。但是需注意的是,主观性的术语应该避免,因为它们可能被错误地解释为暗示与临床效应某种程度的关联性。例如,ADA阳性人群的滴度可以报告为中位数和四分位数范围(IQR),但不宜使用诸如“高”或“低”等词语,因为人们可能错误地认为高滴度的抗体与临床效应相关(即引起不良事件),而低滴度的抗体则不会(即良性)。
图3.ADA滴度动力学。研究中随时间变化的这种滴度图有助于确定ADA水平在治疗过程中是否随时间而变化。每个框图表示滴度范围、Q1、中位数(Q2)、Q3,不包括异常值(星标)。
ADA数据可以酌情以表格、文本或图像等形式显示。其中,以表格形式提供原始数据可以帮助监管机构能够进行独立的分析,以验证所提交的结果。当在表格中提供样本分析结果时,最好包括:受试者识别号、临床站点识别号(姓名或编号)、计划的随访或给药访问(预定时间点)、给药剂量/频率、样本采集日期(实际时间点)、测定的药物血清浓度、样本ADA的状态和滴度、中和能力状态等。ADA阳性受试者数量很少的研究可能会限制某些数据分析的进行。
总结与前瞻
此文为本《蛋白质和多肽药物临床免疫原性的评估和报告》系列的第一篇,包含相关定义和术语、ADA免疫反应的特点以及其临床相关性分析。后续文章将涉及ADA状态与PK/PD,临床安全性和疗效的关系。
特别声明
本文如有疏漏和误读相关指南和数据的地方,请读者评论和指正。所有引用的原始信息和资料均来自已经发表学术期刊、官方网络报道等公开渠道,不涉及任何保密信息。参考文献的选择考虑到多样化但也不可能完备。欢迎读者提供有价值的文献及其评估。
参 考 文 献
1. Guidance for industry: immunogenicityassessment for therapeutic protein products. In: U.S. Department of Health andHuman Services (DHHS), Food and Drug Administration (FDA), Center for DrugEvaluation and Research (CDER), Center for Biologics Evaluation and Research(CBER).http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM338856.pdf2013.Accessed 18 Mar 2014.
2. Shankar G, et al. Assessment andreporting of the clinical immunogenicity of therapeutic proteins and peptides –harmonized terminology and tactical recommendations. AAPS J. 16(4), 658–673(2014).
3. Mire-Sluis AR, et al.immunoassays used in the detection ofhost antibodies against biotechnology products. J. Immunol. Methods 289, 1–16 (2004).
4. Shankar G, et al. Recommendations forthe validation of immunoassays used for detection of host antibodies against biotechnologyproducts. J. Pharm. Biomed. Anal. 48(5), 1267–81 (2008).
5. Smith HW, Moxness M, Marsden R. Summaryof confirmation cut point discussions. AAPS J. 13(2), 227–229 (2011).
6. Swanson JS, Chirmule N. Assessingspecificity for immunogenicity assays. Bioanalysis 1(3), 611–7 (2009).
7. Schellekens H. Bioequivalence and theimmunogenicity of biopharmaceuticals. Nat Rev Drug Discov. 2002;1(6):457–62.
8. Kuus-Reichel K, et al. Willimmunogenicity limit the use, efficacy, and future development of therapeuticmonoclonal antibodies? Clin Diagn Lab Immunol. 1994;1(4):365–72.
9. Koren E, Zuckerman LA, Mire-Sluis AR.Immune responses to therapeutic proteins in humans—clinical significance,assessment and prediction. Curr Pharm Biotechnol. 2002;3(4):349–60.
10. Schellekens H, Casadevall N.Immunogenicity of recombinant human proteins: causes and consequences. JNeurol. 2004;251 Suppl 2:II4–9. doi:10.1007/s00415-004-1202-9.
11. Wolbink GJ, Aarden LA, Dijkmans BA.Dealing with immunogenicity of biologicals: assessment and clinical relevance.Curr Opin Rheumatol. 2009;21(3):211–5.
12. Yanai H, Hanauer SB. Assessing responseand loss of response to biological therapies in IBD. Am J Gastroenterol. 2011;106(4):685–98.
13. Casadevall N, et al. Pure red-cellaplasia and antierythropoietin antibodies in patients treated with recombinanterythropoietin. N Engl J Med. 2002;346(7):469–75.
14. Macdougall IC. Antibody-mediated purered cell aplasia (PRCA): epidemiology, immunogenicity and risks. Nephrol DialTransplant. 2005;20 Suppl 4:iv9–iv15.
15. Schellekens H. Immunogenicity oftherapeutic proteins: clinical implications and future prospects. Clin Ther.2002;24(11):1720–40.
16. Shankar G, Pendley C, Stein KE. Arisk-based bioanalytical strategy for the assessment of antibody immuneresponses against biological drugs. Nat Biotechnol. 2007;25(5):555–561.
17. Koren E, Smith HW, Shores E, Shankar G,Finco-Kent D, Rup B, et al. Recommendations on risk-based strategies fordetection and characterization of antibodies against biotechnology products. JImmunol Methods. 2008;333(1–2):1–9.
18. Ponce R, et al. Immunogenicity ofbiologically-derived therapeutics: assessment and interpretation of nonclinicalsafety studies. Regul Toxicol Pharmacol. 2009;54(2):164–182.
19. Jahn EM, Schneider CK. How tosystematically evaluate immunogenicity of therapeutic proteins—regulatoryconsiderations. New Biotechnol. 2009;25(5):280–286.
20. Shankar G, Devanarayan V, Amaravadi L,Barrett YC, Bowsher R, Finco-Kent D, et al. Recommendations for the validationof immunoassays used for detection of host antibodies against biotechnology products.J Pharm Biomed Anal. 2008;48(5):1267–1281.
21. Buttel IC, et al. Taking immunogenicityassessment of therapeutic proteins to the next level. Biologicals.2011;39(2):100–109.
22. Wang YM, Fang L, Zhou L, Wang J, Ahn HY.A survey of applications of biological products for drug interference ofimmunogenicity assays. Pharm Res. 2012;29(12):3384–3392.
23. Male C, et al. Predictive value ofpersistent versus transient antiphospholipid antibody subtypes for the risk ofthrombotic events in pediatric patients with systemic lupus erythematosus.Blood. 2005;106(13):4152–4158.
24. Chirmule N, Jawa V, Meibohm B.Immunogenicity to therapeutic proteins: impact on PK/PD and efficacy. AAPSJ.2012;14(2):296–302.
关于博济医药 临床研究服务:
博济医药拥有一支规模庞大、专业成熟的临床研究队伍,可提供包括医学、项目管理、监查、稽查、数据管理和统计分析、生物样本检测在内的临床试验全流程解决方案。截至2020年,博济医药服务的客户超1000家,完成800多项临床试验项目,助力客户获得新药证书60多项、生产批件超过80项。在有丰富的临床试验服务经验,服务项目涵盖临床研究各个领域,在肿瘤、肝病、消化等创新药领域拥有独特的临床服务体系。
博济医药在全国设有40多个临床监查网点,与全国近600个临床试验机构展开合作,并运用ORACLE OC/RDC及CTMS系统,控制临床数据采集的及时性、管理临床试验过程的规范性。