









在竞争日益激烈的制药行业,按照法规、指南等传统模式进行生物分析,已经无法适应不断增长的资源、时间、生产效率和加速决策的需求。通过一个分级验证适合其用途的PK LBA方法的方式,可以在生物药开发的早期阶段做出科学合理的决策,从而提高新药开发的效率。
本文将着重介绍并描述PK LBA方法中校准曲线的设计、生成、编辑以及拟合模型和权重选择的相关思考和建议。由于篇幅有限,本文将采用上下篇的形式来展开论述,敬请垂注!
1.导论
校准曲线(Calibration curve)代表了检测到的响应变量与标准参照(比)物(reference standard)浓度之间的关系,这个关系假定了该标准参照物能够代表真实测试样本中的目标分析物(analyte of interest)或待测物。
校准曲线用于通过插值计算定量地测定样本中未知浓度的待测物。校准曲线的产生,是通过将待测物加入被判断为能真实地代表样本的基质中而形成校准品,随后依照未知样本和质量控制样品(QC)的仪器读出值,然后根据校准(或标准)曲线进行内插从而计算未知样本的浓度。
为了优化拟合非线性校准曲线,应当考虑三个因素:
(1)拟合平均浓度与响应的关系;
(2)对非线性剂量-响应曲线中的已知差异率(heteroscedasticity/non-constant response-error relationship,非恒定的响应-误差关系)使用适当的权重;
(3)使用适当的曲线拟合算法来估算拟合的参数。
对样本定量的准确度取决于该分析方法的校准曲线的稳健性和可重复性,该分析方法反过来又取决于参照(比)物(reference material)和其他测试组分(assay component)的性能。应该在方法开发阶段中彻底评估配体结合式测试(LBA)方法的组成成分(即试剂)的性能特性,这些包括但不限于固体或固定的表面(如96孔板)和捕获/检测抗体。同时,应当制定适当的计划,以监控批次间相关试剂的一致性。
此外,相关法规指导文件和该领域专家发表的指导性文件中定义了设计校准曲线的一般要求、校准样品的验收标准以及如何选择适当的回归模型。遵守这些已发布的准则和要求可提高校准曲线的可重复性(跨运行和跨研究)。
总之,每个生物分析实验室都有责任在其标准操作程序(SOPs)中定义LBA校准曲线的设计、产生、验收和编辑标准。本文旨在为产生校准曲线以及处理校准品数据点提供建议和最佳做法。虽然本文的内容也可能适用于某些生物标志物的检测方法,但其重点仍然是用于定量药代动力学(PK)的LBAs的校准曲线。其他所有的测试类型或格式都暂不讨论。
LBAs和色谱分析方法的校准曲线之间存在着关键区别。在LBAs中,仪器响应可能与待测物的浓度呈正比关系(directly),也可能与待测物浓度呈反比关系(inversely),这取决于该分析方法是非竞争性还是竞争式的。无论采用何种格式,可以使用半对数坐标系(semi-log scale)将曲线转换成响应与待测物浓度之间的S型关系。这与色谱分析方法不同,在色谱分析中,仪器响应通常是浓度的线性函数,两者在大多数校准曲线范围内是呈成正比例关系的。对于色谱法来说,线性关系的丧失表明该方法已经达到了它的检测极限。
LBAs方法依赖于待测物与结合试剂(如抗体或受体部分)的相互作用,而这与传统的色谱分析不同,在传统的色谱分析中,待测物的检测不依赖于其与某个大分子的结合(binding)。蛋白质-蛋白质相互作用的动态平衡性质导致了LBAs的非线性响应。此外,由于LBAs的效能在很大程度上取决于其所使用的生物试剂的性能,因此其检测结果往往表现出较大的变异性(variance)。
非线性的LBA校准曲线限制了曲线上端和下端的浓度-响应的相关性,使得曲线处于平台状态,因此形成S型曲线。在典型的sigmoidal校准曲线中,下部平台(渐近线asymptote)及其附近代表了背景响应,上部平台代表接近最大响应。通常认为4PL模型是拟合此形状校准曲线的首选。另外,在校准曲线的渐近线(上、下平台部分)上进行定量将导致产生较差的精密度和准确度。这些特性最终缩小了LBA方法的有效定量范围,使得选择合适的非线性数据拟合算法变得更加重要。
有多种商业软件可用来执行LBAs的非线性回归。大多数分析仪器制造商均有提供与设备兼容的非线性回归软件。实验室可选择安装独立软件或使用其实验室信息管理系统(LIMS)来进行回归拟合,具体取决于哪种软件更能满足其需要和要求。
用于拟合LBA校准曲线的软件应该具有使用4个和5个参数logistic模型(Four and five parameter logistic regression,4和5 PL)进行回归分析的能力,并且能够:
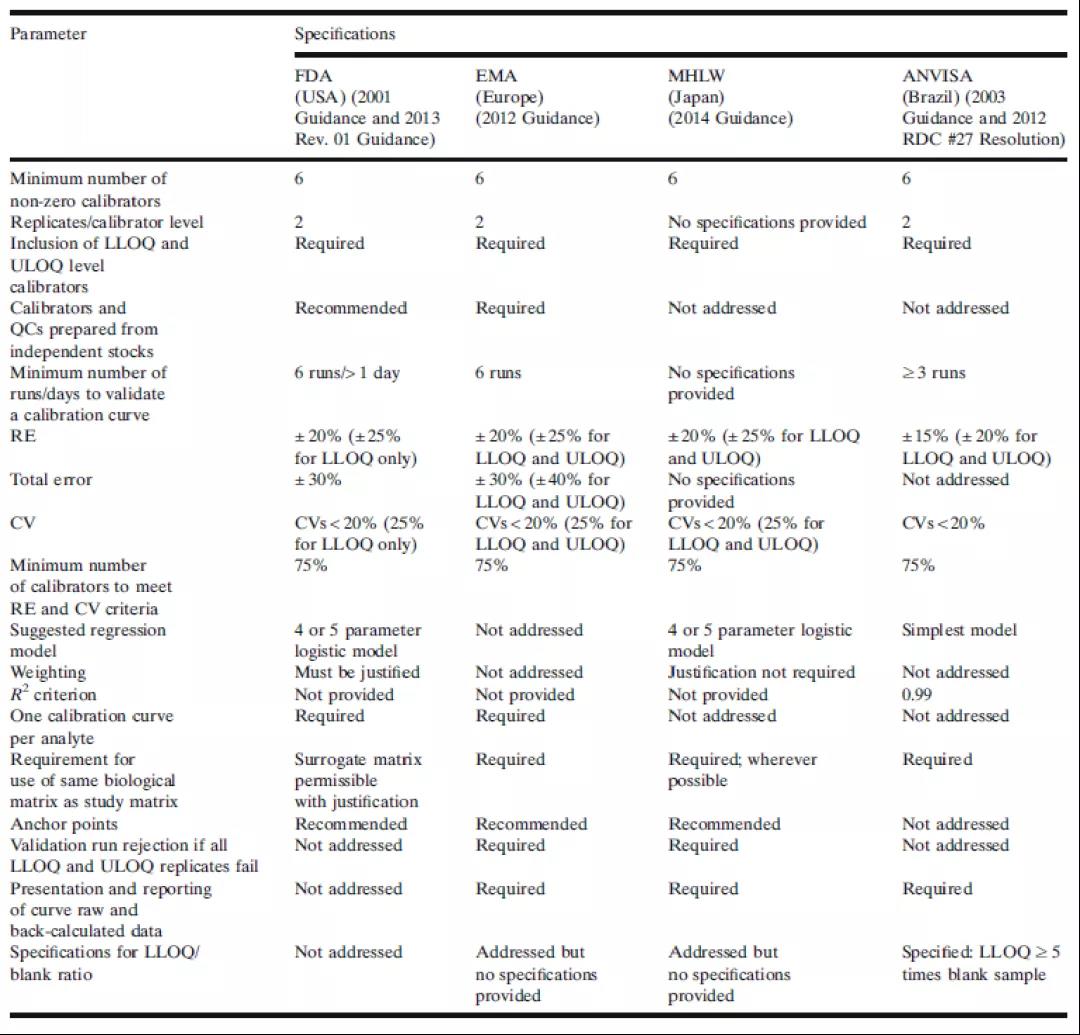
当前,一些生物分析指南或法规文件的生物分析部分已经对校准标准曲线的最低要求进行了确定。这些指导性文件对LBA曲线的需求和性能预期通常是相互一致的。表1总结了来自美国食品和药物管理局(FDA)、欧洲药品管理局(EMA)、日本卫生、劳动和福利部(MHLW)和巴西卫生监管机构(ANVISA)对校准曲线的要求。FDA和EMA指南是绝大多数生物分析实验室遵从的主要指南。各个制药厂商应根据各自计划所提交的新药审核监管机构来决定自身需要遵从的指南法规。
校准标准品(Calibrator standards)是通过将已知浓度的参照(比)药物加入到与研究样本基质相同或一致的,经过认证的基质(matrix)中而产生的。这些校准标准品的浓度——响应关系则确定了该测试方法的校准曲线。在涉及多种药物联合使用的研究中,研究样本中的每个待测物都需要一条校准曲线。校准标准品的制备必须独立于质量控制样品(QCs),以避免潜在的药物加入造成误差的扩散或放大。
这意味着校准标准品和QCs可能不能使用相同中间库存的药物参照(比)品来制备,但可以通过连续稀释药物参照(比)品的初级或中级库存来制备校准标准品。制备校准标准品不需要在每个浓度水平上单独外加药物参照(比)品来制备,尽管这种做法意味着需在更高一级的水平上对校准曲线的质量控制,并可以监测外加药物参照(比)品的精确度。无论药物参照(比)品的中间库存组成如何,校准标准品应包含至少95%的研究样本基质。
使用LBAs方法的期望是在可能的情况下,尽一切努力在生物基质中制备校准标准品,该生物基质在物种、组成成份和基质预处理等方面必须与研究样本的基质相匹配。例如,若研究样本是未经过滤的血清,那么制备校准标准品的基质也必须是来自相同物种的未经过滤的血清。需要注意的是,研究人群通常是伴有相关的疾病,而校准品基质(calibrator matrix)则是来自于健康的受试者。如果没有其他对待测物做定量分析的方法,那么用替代基质来制备校准品也是合理的,例如当研究使用的基质稀缺或很难获得时,当研究药物有内源性同源物时,在对生物标志物进行检测时等。
当使用替代基质来制备校准品时,该生物分析方法应该使用研究样本基质制备的QCs和研究基质选择性样本(study matrix selectivity sample)进行验证,并与替代基质中制备的校准曲线进行比较和评估。除此之外,还建议分析者进行其他试验,以证明替代基质与研究基质的稀释曲线两者之间的可比性。一种普遍接受的方法是测试上下渐近线、增长率(growth rate)以及5PL曲线中不对称因子的等同性。当前业界内已提出如何测试平行性(parallelism)的方法,平行性测试是一个评估LBA方法相对准确性的基本实验。通过分析稀释对基质中内源性待测物定量的影响,可以在单个实验中评估该测试方法的选择性、基质效应、所需的最小稀释率、健康和患病人群的内源性待测物的水平以及LLOQ。
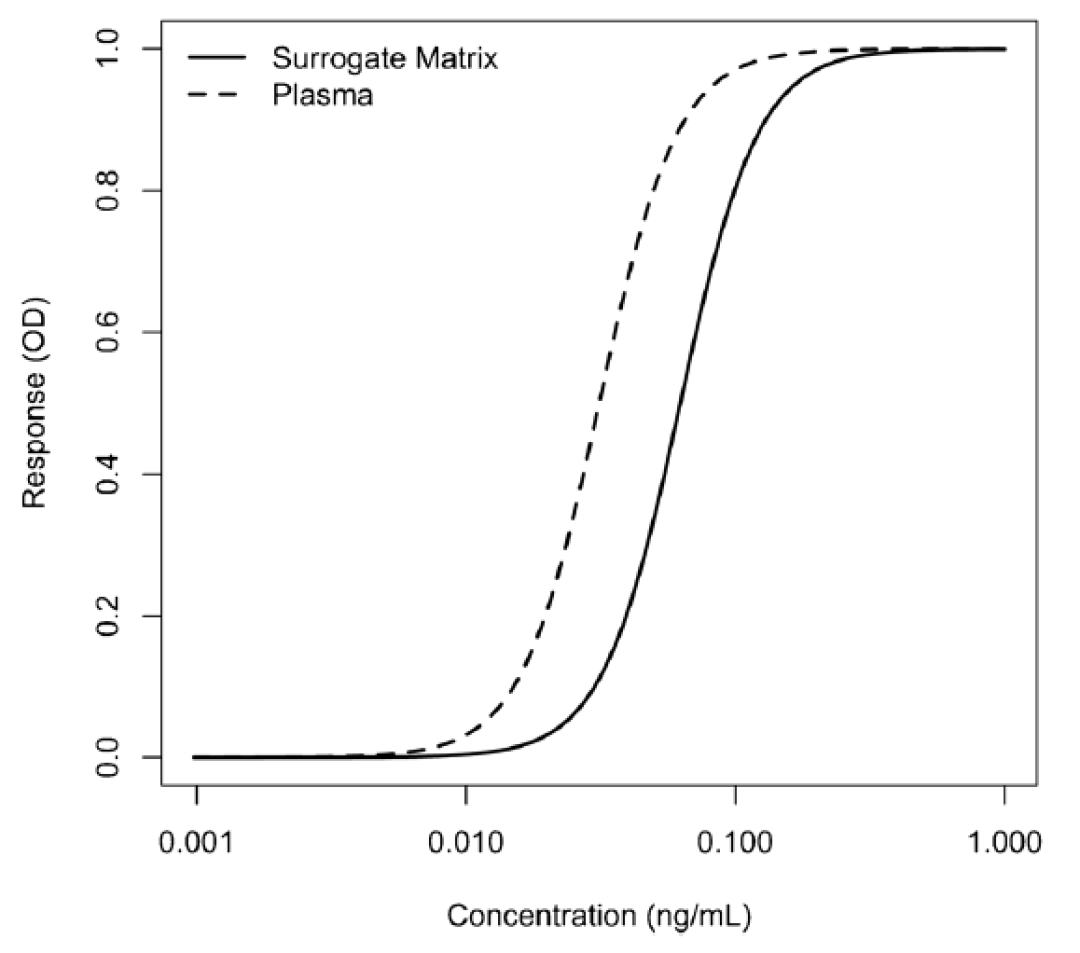
这些等效性测试的接受标准尚未确定,但已在业界广泛讨论。图1提供了在人血浆中制备的校准曲线的示例,同时也提供了在缓冲液(替代基质)中制备的校准曲线。它们是两条平行的曲线。
校准样品可用100%或以最低稀释度(minimum required dilution,MRD)稀释后的基质配制。使用MRD稀释基质的方法的例子包括但不限于罕见基质或在自动化平台上执行的方法,在这些平台上使用100%基质可能会由于有限的可及性(limited availability)或由于基质的粘度而成为问题。在方法开发过程中,应根据测试方法性能来决定校准样品是用100%的基质还是用MRD稀释的基质制备,应根据具体的实际情况进行适当的评估,以确保校准样品和QC的回收率在理论值的预期范围内(例如,±20%)。当在100%基质中制备时,校准样品需要以用于QCs和研究样本相同的MRD来稀释。未知样品回算的浓度应报告为100%基质中的浓度。
在生物分析应用中,选择认证后的混合基质(qualified matrix pool,QMP)的过程至关重要。建议认证和储存足够的混合基质,确保足以用来完成方法开发、验证和至少第一次研究中样本分析。还建议将QMP储存在与QCs和研究样本相同的条件下。例如,如果样本的储存温度小于或等于-65℃,则QMP也应储存在该温度范围内。
在方法开发过程中,可以筛查、选择和合并单个基质样本或单个混合基质来生成QMP,商业来源的混合基质也可能适用。QMP筛查应包括评估对未外加和外加了待测物的基质样品的检测信号。例如,出于筛查的目的,可以在单个基质样本加入相当于LLOQ水平的待测物。具有高背景或加标回收率不合格的基质样品应排除在QMP之外。基质样品的验收标准的一个实例可以是:使用QMP制备的校准曲线,至少有80%的所有单个基质样品的加标回收率在理论浓度的80%至120%之间。QMP应该能代表研究人群,通过混合满足接受标准的个体基质样本来制备。
LBA校准样品可以是新鲜制备的或者冷冻的。不少实验室在所有阶段习惯使用新制备的校准样品,包括方法开发、研究前验证和随后的生物分析。新鲜的校准样品可以从原始或中间参考标准的库存(original or intermediate reference standard stock)中调取。如果使用中间参考标准,必须在发布验证报告之前确定其稳定性。在研究前的方法验证中,使用新制备的校准样品来评估冷冻的QCs,可以初步建立QCs的稳定性。
一旦初步确立了QC的稳定性,实验室就可以利用这些信息来制备和储/冻存单个校准样品。如果将LLOQ和ULOQ样品包含在稳定性测试中,且稳定性测试窗口覆盖了校准样品的存储期,则此方法就是可以接受的。预先认证和冻存校准样品的目的是减少测试运行之间的变异性和提高操作效率。冻存的校准样品应分装在足够一次性使用的容器中,并应避免校准样品的反复融冻。
监管机构为校准曲线的设计提供了指南。以下是指南准则的简短总结:
ULOQ和LLOQ分别代表定量分析范围的上限和下限,必须作为研究前验证的一部分进行验证。为了验证LLOQ和ULOQ校准样品,仅仅在这些级别上包括校准样品是不够的,还需要在LLOQ和ULOQ水平上制备验证样品(QCs),这些QCs必须包括在研究前验证的准确度和精密度的评估中。ULOQ校准样品必须分别满足相对误差(RE)为±20%和变异系数(CV)为≤20%的接受标准;LLOQ校准样品则必须分别满足FDA指南中规定的±25%的RE和≤25%的CV,才能被纳入校准曲线。一旦经过验证,LLOQ和ULOQ水平校准样品将成为校准曲线的一个组成部分,并且必须包括在每次测试运行中。
研究中每个待测物在每次分析测试运行应当有一条校准曲线,校准范围必须适合并符合研究样品中待测物的预期浓度范围。一般来说,宽大的定量范围有助于一次性地定量分析含有大范围待测物浓度的大量样本。狭窄的定量范围则限制了该测试方法的分析能力,导致不必要的重复分析,即需要额外稀释从而使样本浓度进入校准曲线范围。虽然将待测物浓度为零的校准样品定义为不含待测物的基质样品不是必须的,但可能是有益的。
目前的建议是对LBA校准样品进行重复(duplicate)分析,尽管变化趋势如高CVs,可能需要进行三次(triplicate)复测。只有在该方法的量化范围内证明了原始响应的稳健性和高精密度的情况下,才能认为使用单次测量(单孔singlicate)是合理的。
2012年EMA生物分析指南中讨论了非线性回归拟合时锚定点的作用,并在整个行业中推荐使用锚定点。锚定点被定义为在测试方法的定量范围之上和之下的校准样品,但它们不受与校准曲线点相同的性能要求的约束。锚定点的价值和在回归分析时使用锚定点并不是被普遍接受的,但是建议将它们作为方法开发过程的一部分,并评估它们对改善整体回归分析的作用。
使用锚定点是否能改善曲线拟合,应基于所提出的数学算法或加权因子,并应在个案基础上确定。锚定点可能特别有助于增强曲线拟合,不仅在过度宽大或狭窄的校准曲线的情况下,而且在使用加权因子的校准模型中也是如此。在某些情况下,位于LLOQ以下的锚定点增强了曲线的拟合,并有助于LLOQ满足其接受标准。
目前监管机构尚未就校准样品之间的间距和ULOQ/LLOQ信号比作出明文指导。一般推荐使用相等的校准样品的间距,如采用对数刻度(例如,on a logarithmic scale of the power of 2),这有利于改善测试方法的性能。
校准样品浓度的选择部分是基于以下的实际考虑:即在进行多个测试运行时,易于制备和无差错地复制校准品浓度。建议分析者使用系列稀释(serial dilution)的方法,并使用一个固定的稀释比例(使用与被分析的实际样本相同的基质稀释校准品)。最终结果是,校准品在浓度范围的对数刻度上的间隔大致均匀/相等的。在这些条件下,Rocke和Jones确定了校准品的最佳稀释比例,该比例通常为2:1或3:1。2:1的系列稀释与6个校准品浓度将产生一系列如1X,2X,4X,8X,16X,32X(稀释倍比为2n: n = 0,1,2,3,4,5的曲线)。对于典型的响应递减曲线(D<A),Rocke和Jones的研究也显示,如果使用最佳间距,校准品浓度曲线的中点应略高于预期的IC50浓度(当LBA用于活性测定时)。这意味着在曲线较小方差(variance)的区域有更多的校准点。对于上述2:1的稀释比例,应调整X,以便所预期的IC50 浓度位于8X稀释的校准浓度点附近。可以添加额外的校准样品(如果已包括在回归模型的评估中)来更好地定义校准曲线的拐点(inflection point)。
在LLOQ附近加入更紧密相邻的校准样品可能有助于在LLOQ失效时,将灵敏度的损失降到最低。在校准曲线中包含零浓度的校准样品不是监管机构的要求或标准做法,然而,如果一个实验室选择将零作为校准曲线拟合的一部分,则建议在LLOQ和零校准样品之间留有足够的间距,以保护LLOQ不会失败。例如,2012年的EMA指南规定LLOQ的信号至少是空白样本信号的5倍。每个实验室应根据测试方法的效能(performance)为其确定适当的间距。
ULOQ/LLOQ浓度比受测试格式(format)、平台(platform)和试剂特性的影响。实验室的目标应该是最大限度地提高ULOQ/LLOQ浓度比,并可设定其最低目标,如10/1。
选择一个合适的非线性回归模型需要经过多次迭代才能获得LBA校准曲线的最佳拟合。目前的监管指南是建议使用能够获得足够拟合度的最简单模型来进行拟合,建议在评估任何可能的权重之前,首先选择回归模型。评估权重能否减轻不同浓度下测试响应不等同的变异性也很重要。
此外,还需要考虑的一个关键参数是可报告的测试结果的质量,对它的考虑应该超越对模型拟合质量的考虑,并且可以通过准确度进行评估。因为准确度概要(Accuracy profile)有助于各种拟合模型的可视化。必须强调的是,目前的研究文献不鼓励如下操作:(1)使用线性函数来近似S形曲线;(2)对数据进行对数-对数转换(log-log transformation)来使得固有的非线性关系近似于线性。
校准曲线常用的非线性模型是4PL和5PL,可以通过许多自动化软件实现拟合。虽然可以使用其他的非线性模型,但须谨慎且应对4PL和5PL模型不适用的特殊情况加以证明。
4PL模型最常见的参数化形式是:
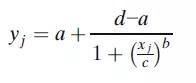
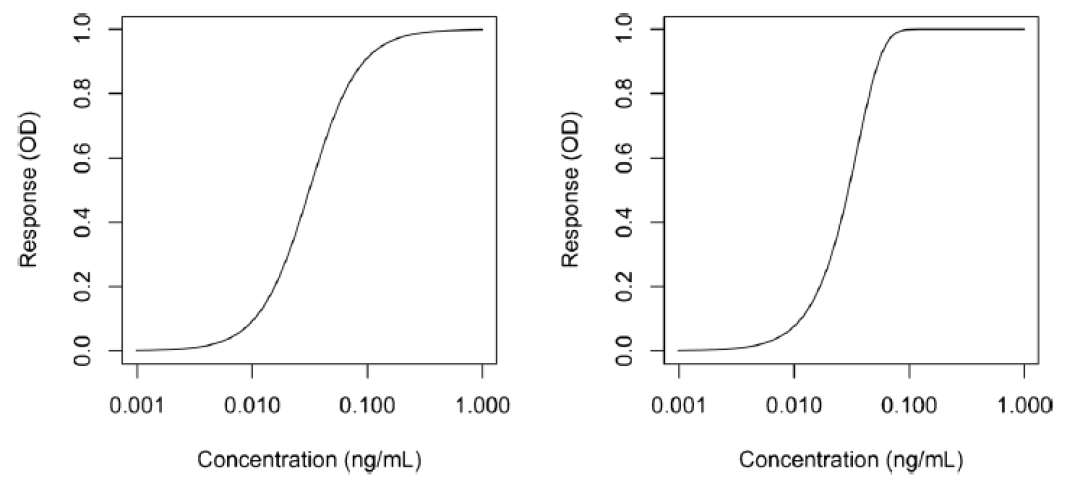
其中yj是对应浓度xj的响应,a为上渐近线,d为下渐近线,c为曲线拐点处的浓度,b为生长因子。该模型的一个特点是拐点附近的对称性,而拐点对应于d和a之间距离的一半。这个模型虽然也可应用,但它通常产生的是一个不对称的校准曲线,因而需要使用5PL模型拟合。
5PL模型的一般参数化形式是:
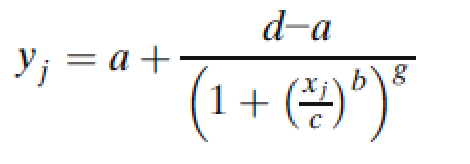
其中g是不对称因子。当g=1时,5PL函数与4PL函数完全等价。图2说明了4PL和5PL曲线之间的差异。
LBA校准曲线拟合的另一个常见挑战是不同校准样品浓度下响应的不相等变异性(unequal variability),即响应的噪声(variance)通常不是恒定的,而是随着响应而变化,这种现象被称为异方差性(heteroscedasticity),可以通过对模型进行加权处理,使其与浓度范围内响应的可变性成比例。如果应对异方差性得当,校准曲线拟合的质量则可以提高。应对的基本思想是在表现出较高变异的响应上减少其"权重"。换句话说,校准曲线与低变异数据更紧密地拟合,并允许稍微偏离较高变异的数据,以这种方式加权可得到更宽泛的定量范围,并在该范围内提高准确度和精密度。如果不使用适当的权重,将会导致LLOQ和ULOQ附近的插值有更大的偏差(bias)和不精密度(imprecision)。适当使用权重可以减少重复测试的响应值中的不相等方差(unequal variance)。
大多数软件都具有执行加权回归所需的功能。软件通常会选择常用的权重,如1/Y和1/Y2,并通过评估响应-误差关系(response-error relation)来选择最佳的权重函数。1/Y的权重增强了曲线底部的点,1/Y2的权重增强了曲线顶部和底部的点。还有其他权重因子和加权方法也同样可以使用,并可能能够更好地满足特定测试方法的需求。
虽然曲线的非对称性和异方差性(heteroscedasticity)是曲线拟合的重要考虑因素,但如果没有适当的预评估,将它们包含在模型中可能会导致拟合一个过于复杂的模型。在校准曲线中包含反映测试方法自然变化的参数可能会导致回算误差和增加所报告的结果的偏差(bias)。
做回归分析时,有许多不同的方法可用于统计评估模型拟合的质量。(包括R2和均方误差(root mean square error)。但这些参数并非适用于所有情况,它们的设计目的是将响应的误差最小化,而不是将所报告的结果的误差最小化。此外,Anscombe’s quartet表明,高度不同的数据可以获得相似的拟合质量,但并不能保证反向预测的相似。准确度曲线(accuracy profile)是一种基于未来可报告结果的替代性图形方法,它将各浓度水平下测试所得的可报告结果与真实值之间相对差异的b-期望容差区间(b-expectation tolerance interval)联系起来。
b-期望容差区间被定义为某个比例(b%)的未来结果预期会下降的区间。该工具可以方便地可视化偏差、精密度和定量限(limit of quantitation,LOQ/定量限,在这里准确度超出了可接受的范围),提供了一种可以用来比较多个模型的简单方法,并方便从中选择精度最高的可报告结果的模型。
图4给出了两条不同的校准曲线及其基于15%相对误差的相关准确度曲线(RE,定义为实验值和理论值之间的差值与理论值之比)的接受极限(acceptance limit)。15%相对误差的接受极限是指南推荐的。
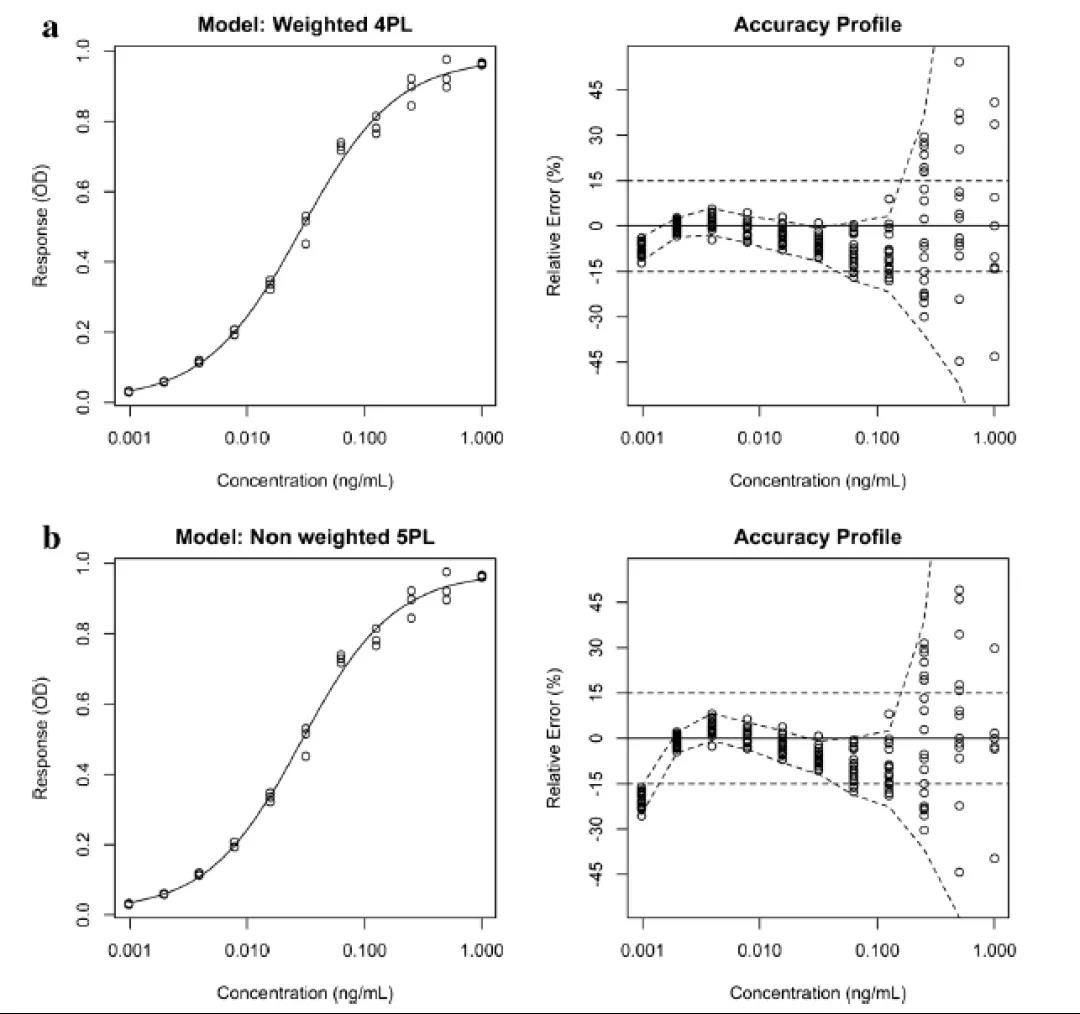
至少有75%的校准样品,包括LLOQ和ULOQ校准样品,按回算浓度,必须通过接受标准:即该回算浓度在理论浓度的±20%(低定量限为±25%)的范围内,而CVs低于20%(在低定量限为<<>25%)(另见表1)。对于每个校准样品,应该报告回算浓度的CVs,而不是仪器响应的CVs。首先,应该根据精密度排除校准样品。每次排除后,应重新对曲线进行回归分析和评价。其次,应该一次只排除一个校准样品,并且按偏差大小的顺序,从拥有最高偏差(RE)的校准样品开始。另外,只有在需要时才排除其他校准样品。
屏蔽(masking)被定义为:在标准曲线回归时不使用某个校准点,而该校准点的数值仍存在于系统中。在讨论曲线编辑时,屏蔽和排除(exclusion)这两个术语可以互换使用。每个A校准点通常是在微孔板的两个孔(复孔)中进行重复(duplicate)测试。如果曲线拟合是基于复孔测试的平均值,则只有当两个孔的测试结果都满足接受标准时,才能接受该校准点。在这种情况下,不建议排除一个孔的数值,而只使用另一个孔的结果。在某些试平台,如Singulex,每个校准点可能测试三次,在这种情况下,三个孔中至少有两个必须通过,才能接受该校准点。
如果LLOQ和ULOQ校准点的所有重复测试都失败,那么该验证运行就失败了,应该调查失败的可能原因(EMA 2012)。编辑校准曲线只能在可以确定失败原因的情况下进行,例如:记录在案的外加药物误差,或移液器误差,或使用了先验的统计标准。校准曲线的编辑必须独立于QC的评估, 这意味着,不能排除校准点以方便QC的通过,除非不符合与校准样品相关的接受标准。每个实验室必须在其标准操作程序中定义如何屏蔽和编辑校准点。
屏蔽和排除校准样品的一般准则如下:
一旦测试运行通过,每个样本都可以根据校准曲线进行评估。如果一个未知样本结果的CV%可接受,并且平均浓度落在该方法的定量范围内,则该结果可以被报告。如果样本的平均浓度超出了定量范围,则应在适当的稀释后重新测试该样本,以获得在定量范围内的结果。如果一个测试结果在定量范围内,而另一个复测结果低于LLOQ或高于ULOQ,则应该报告平均值,前提是平均值在经过验证的定量范围内,且CV%是可接受的。样本浓度结果应报告为100%的基质,在考虑了测试方法的MRD和任何其它稀释的倍数之后。
正如在前一节中提到的,如果LLOQ校准点失败且必须被屏蔽,定量范围就截断到下一个最低的校准点。在这种情况下,LQC仍必须由可接受的校准样品包含在内(bracketed),否则检测失败。如果是ULOQ失败,曲线的上端向下移动到下一个可接受的校准点,且必须包含在可接受的校准样品内,否则检测也同样失败。如果样本的测定值低于LLOQ或高于ULOQ,则必须调整样本的稀释倍数,再重新分析该样品,使其测定值落在定量范围内。
LBA校准曲线可能容易随时间发生校准漂移。校准曲线性能的漂移定义为由于参比标准品、分析试剂和其他测试组分的反应性或结合特性的变化而导致的校准偏移。这种变化可能会改变校准曲线的斜率或其他性质,最终导致样本的浓度报告为过高或过低。同时,该方法的定量上限和下限,或样本的稀释线性,也可能发生变化。
可能导致校准曲线性能漂移的因素包括但不限于:(a)新批次的混合基质;(b)关键试剂特性(如纯度、特异性和捕获或检测抗体的结合亲和力)的变化;(c)含有蛋白质或脂质添加剂或载体的非关键试剂的性能变化;以及(d)对参比标准品配方的修改。应在方法开发的早期就开始监测校准曲线的性能,并在研究前验证和样本分析阶段继续进行。此外,在多个临床研究的时间跨度上,跟踪校准漂移是至关重要的。但是,目前还没有用于监测校准曲线性能的共识或已经建立的方法。
监测校准曲线漂移的建议包括:
旨在确定药物浓度以支持药代动力学研究的,高质量和可靠的配体结合测试方法(LBA)在生物药的开发过程中起着至关重要的作用。在典型的LBA方法中,使用校准曲线通过内插值(interpolate)测定质量控制样品和未知样品中的药物浓度。对大多数LBA方法,预计测试信号与药物浓度的关系是非线性的。因此,建议应用多参数,通常为4或5参数Logistic regression (4PL或5PL)的数学方法,在定量范围内,拟合至少6个校准样品的数据点以获得校准曲线。可考虑使用其他校准样品(包括零点或其他锚定点)来提高曲线拟合的质量。
应根据对准确度曲线(accuracy profile)的分析,选择最适当和最简单的回归模型(可能应用加权因子,weighting factor)的相关回归分析的软件,作为实验室信息管理系统的一部分,或作为仪器数据分析包的一部分,或作为独立软件包都可以提供这样的数据分析。校准曲线的质量以及最终测试结果的质量高度地依赖于校准样品的制备过程、所使用的样本基质的类型以及校准样品的储存条件。FDA、EMA、MHLW和ANVISA发布的机构指南在不同程度上讨论了校准曲线参数及其性能要求,其中许多要求是相互一致的,但也存在一些差异。
本文旨在描述进行回归模型选择、校准曲线设计和数据分析的方法,主要目的是帮助读者开发高质量的生物药定量分析方法,以支持非临床和临床药代动力学研究。文中花了较大的笔墨详细讨论的一个重要内容是与编辑校准曲线的规范和适当规则有关。同时提供了对于编辑校准曲线的建议,这些建议遵循科学上健全,同时也符合行业惯例的原则。当然,其他方法也可以,如果其科学性和适用性被证明是可以接受的。
本文如有疏漏和误读相关指南和数据的地方,请读者评论和指正。所有引用的原始信息和资料均来自已经发表学术期刊、官方网络报道等公开渠道, 不涉及任何保密信息。参考文献的选择考虑到多样化但也不可能完备。欢迎读者提供有价值的文献及其评估。
19.U.S. Food and Drug Administration, Title 21 of the U.S. Code of Federal Regulations: 21 CFR 11 “Electronic Records; Electronic Signatures”, Aug 2003.https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm.














